Yves here. KLG unpacks a recent overview paper on SARS-CoV-2 mechanisms. Although this discussion is, of necessity, somewhat technical, Covid isn’t going away any time soon. So making an investment in understanding it better should help, if nothing else in discerning whether various commentators know what they are talking about.
By KLG, who has held research and academic positions in three US medical schools since 1995 and is currently Professor of Biochemistry and Associate Dean. He has performed and directed research on protein structure, function, and evolution; cell adhesion and motility; the mechanism of viral fusion proteins; and assembly of the vertebrate heart. He has served on national review panels of both public and private funding agencies, and his research and that of his students has been funded by the American Heart Association, American Cancer Society, and National Institutes of Health
We are 40 months into the COVID-19 pandemic, and we still do not have a good handle on how SARS-CoV-2 causes disease. Yes, the clinical correlates of COVID-19 are well described if somewhat less well understood, especially the nature of long COVID. Clinical management of disease has improved, and the pandemic has been declared over by the President of the United States. Nevertheless, several hundred Americans die every day of COVID-19, and this is undoubtedly an underestimate. Various interested parties argue about the significance of excess mortality over the past several years, but there is little real doubt about the cause.
Even so, the molecular and cellular causes of COVID-19 disease have not been described well enough for virologists and clinicians to have a sure grasp of what they, and we, are dealing with. Given that the response of the scientific community (>340,000 papers in PubMed with “COVID” as the query as of 11 March 2023) has been so large, this seems a bit strange (1). Nevertheless, a paper published in Nature Cell Biology on 9 March 2023 has made what is likely to be real progress: SARS-CoV-2 infection induces DNA damage, through CHK1 degradation and impaired 53BP1 recruitment, and cellular senescence (Open Access).
Although that is a very technical title, the data are presented very clearly and completely. The objective of this post is to guide the reader through representative experiments and their interpretation. I believe this is important because the explosion of COVID-19 literature has been so overwhelming. A lot has been published, and some of it has been hurried. More of it has been more “sciency” or “scientistic” than truly scientific. Fashion in biomedical science is certainly a thing (a subject for another time). This paper is one of the best I have read on the molecular and cellular derangements associated with COVID-19.
All that is really needed to appreciate the results in this paper is some high school biology:
- According to the Central Dogma of Molecular Biology, DNA makes RNA makes protein, and that proteins are the working parts of the process and virtually all other cellular functions. DNA is a double helix of A-T and G-C base pairs.
- All cells have mechanisms to ensure that DNA damage is repaired before cell division is completed, and that when maintenance of genome integrity is disrupted normal cellular functions will fail and this failure results in disease.
- Viruses are “organisms” that replicate only when they infect host cells and viral proteins allow the virus to hijack normal cellular processes to produce new virus particles. A pathogenic virus causes disease as a consequence of its life cycle or the activity of one or more of its proteins. Common viral diseases, historical and current, include polio, smallpox, AIDS, various conditions caused by herpes viruses, cancer (e.g., cervical and head/neck cancer caused by HPV, human papilloma virus), and COVID-19. Virus genetic material can be RNA or DNA.
Understanding this paper requires some guidance on the interpretation of the data. This is really no more difficult than understanding why Silicon Valley Bank failed last week. I am sure by the time this post appears we will both know more about the causes and consequences and responses of that untoward event. (As it turns out on Monday evening, 13 March 2023: “Nothing to see here, move along” seems to be a common trope among the more self-interested parties). The COVID-19 biology described here is certainly no more difficult to understand than “private equity,” derivatives, and credit default swaps, which are also maledictions that afflict us all in one way or another.
The coronavirus SARS-CoV-2 has a small RNA genome of 30 kb (30,000 RNA nucleotides in one single RNA molecule (2). This genome encodes 26 proteins, including 16 non-structural proteins (NSPs), 4 structural proteinssuch as the nucleocapsid (N) protein and 6 accessory proteins; the Spike protein of SARS-CoV-2 is the antigen for the COVID-19 mRNA vaccines. The DNA Damage Response (DDR) is a network of essential pathways that sense DNA lesions, signal their presence, and coordinate repair of the DNA before cell division can proceed without the mutations that would otherwise result if genome stability were not maintained. These DNA lesions include single-strand and double-strand breaks (SSBs and DSBs) in DNA, which are detected by various proteins in the cell. Some research has been done on DNA viruses and DDR, but less is known about RNA viruses. While it has been suggested that SARS-CoV-2 infection affects host cell DDR machinery, not much was known about DDR and RNA viruses before this paper was published last week (9 March 2023). Two proteins encoded in the SARS-CoV-2 genome are the culprits in interference with the DDR. They are ORF6 (“open reading frame” designates a protein sequence of previously unknown function that has been identified in a DNA or RNA sequence) and NSP13.
Paraphrased from the Introduction:
SARS-CoV-2 infection causes DNA damage and activation of an altered DDR. DNA damage is the consequence of the degradation of the cellular protein CHK1 by ORF6 and NSP13…Depletion of CHK1 leads to a shortage of the building blocks of DNA (the four building blocks – dNTPs – of DNA: dATP, dGTP, dCTP, dTTP; or A-T and G-C base pairs of the DNA double helix). This deficiency results in impaired cell division, DNA damage accumulation, DDR activation, induction of inflammatory pathways and cellular senescence. Supplementation with precursors of dNTPs is sufficient to counteract this cascade of events by allowing DNA replication to proceed. SARS-CoV-2 N-protein also interferes with DNA repair. In addition to the cultured human cells used as an experimental model, these events also occur in mice infected by SARS-CoV-2 and in patients with COVID-19.
Thus, this paper has identified several molecular concomitants of inflammation and associated cellular pathologies well known to afflict COVID-19 patients. In what follows I will illustrate some of the what and the why of these conclusions. The data look like cuneiform tablets at first glance, but only a few keys are necessary to understand them. This will be useful to interested readers who want to go deeper into the results of this paper, which is freely available and well worth the effort if productive rabbit holes are alluring. In my view, this is an exemplary scientific publication and, under the circumstances of COVID uncertainty, much needed. This primer will also make it possible to go beyond the Abstract and Conclusions of any other COVID-19 paper that concentrates on the molecular and cell biology of COVID-19. The devil is in the details, which have been devilishly lacking in too much of the extensive COVID-19 literature.
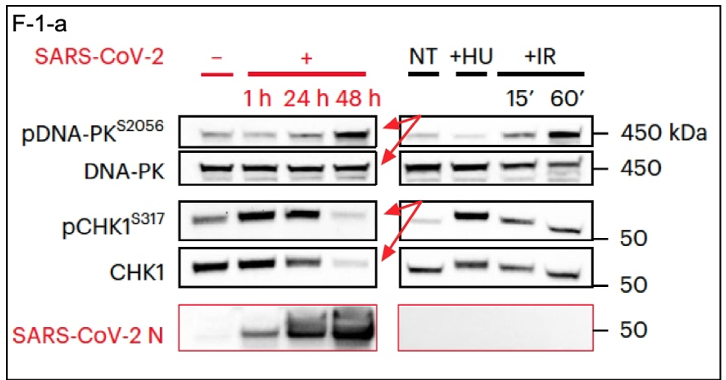
SARS-CoV-2 infection activates the DDR cascade and leads to CHK1 deficiency, which interferes with DDR (Figure 1a, paper). Controls are in the first of the four lanes, i.e., no SARS-CoV-2. This represents the basal or normal state of these cells [Huh7, a human liver cell line (3) naturally permissive for SARS-CoV-2 infection)] in the absence of SARS-CoV-2. Experimental samples were taken at 1, 24, and 48 hours post-SARS-CoV-2 infection. The top panel in the excerpt below (F-1-a) shows DNA-PK, which is a master regulator of DDR that must be activated by having a phosphoryl group added to it, denoted by pDNA-PK(S2056) (4). These two panels show that DNA-PK levels remain the same from time zero through 48 hours of infection but that the active phosphorylated version, i.e., pDNA-PK(S2056), has increased (red arrows). At the same time, the inverse is seen with CHK1 (red arrows). Both the protein and its active phosphorylated version are severely depleted at 48 hours, with this result: No CHK1, no downstream functional DDR despite the activation of the master regulator DNA-PK. Similar results are seen in the right panels with treatment by hydroxyurea or ionizing radiation, both of which disrupt DNA replication, although the response with CHK1 is not as strong as with SARS-CoV-2 infection. The bottom panel shows that SARS-CoV-2 N-protein increased during the experimental time course due to a productive infection with the virus. Taken together, the data presented here and in other experiments described are basically as good as they get.
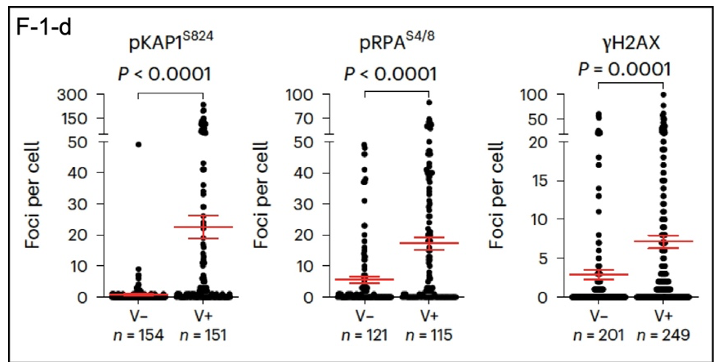
What happens in individual cells? DDR markers can be identified using imaging of proteins tagged with fluorescent reporters in infected cells; DAPI stains DNA (Figure 1c; in the paper). The fluorescence images are best viewed enlarged in the original, so I will concentrate here on quantitative data extracted from the images. These results are presented below (F-1-d). Infection with SARS-CoV-2 (V+) increases the number of DDR foci in infected cells, as identified by phosphorylated DDR proteins pKAP1, pRPA, and gamma-H2AX, with significantly more positives in virus-infected cells. Another significant result shown here is the variability of the effects of the virus on individual cells. This may be reflected in the variabilityof the course of disease in individual patients.
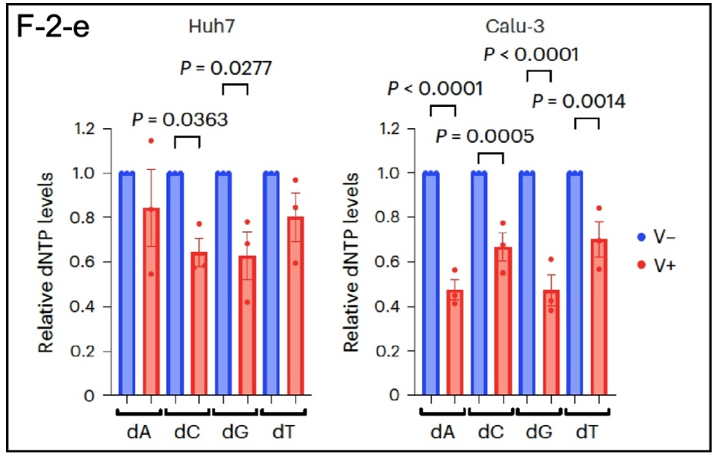
As shown in F-2-e, SARS-CoV-2 also reduces RRM2 levels (5), which leads to a shortage of the precursors for DNA synthesis (dATP, dCTP, dGTP, and dTTP), which are reduced by as much as 50% in Huh7 cells (liver) and Calu-3 cells (lung adenocarcinoma). This perturbation interfered with normal cell division and made it difficult for cells to proliferate. In the organism this leads to DNA damage and the secretion of inflammatory cytokines. Systemic inflammation is a major consequence of SARS-CoV-2 infection.
Which SARS-CoV-2 proteins are the culprits in these cellular perturbations? The authors of this paper individually expressed 24 of the 26 viral proteins in Huh7 cells using conventional techniques. Expression of both ORF6 and NSP13 reduced CHK1 and RRM2 protein levels by
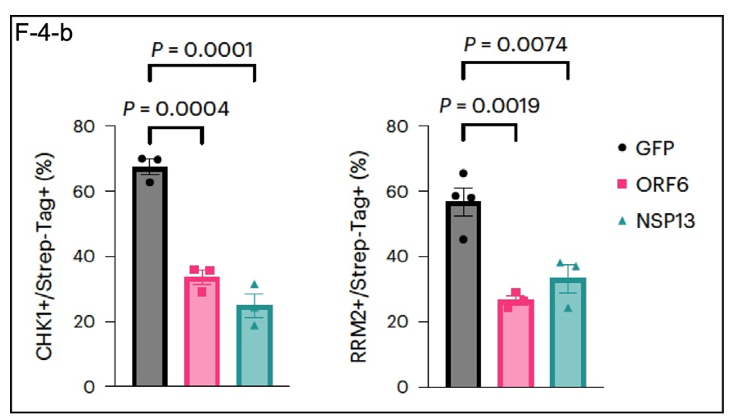
~50% (F-4-b). GFP (described here) as an exogenous protein control, so this result is not likely to be due to a general effect on protein expression by the viral proteins ORF6 and NSP13.
As mentioned in endnote (3), this kind of research on cultured cells is useful, but whether the cells are derived from tumors or immortalized by transformation with a tumor gene or cultured as normal primary lung or liver cells from the organism, they do not recapitulate the organism. Figure 7 in the paper (excerpt not shown here) shows that SARS-CoV-2 infection causes DNA damage in the lungs of mice expressing the human ACE2 protein along with expression patterns of DDR proteins expected from the experiments on cultured cells. ACE2 is the “receptor” (6) for SARS-CoV-2. As expected, CHK1 and RRM2 levels decreased in these “humanized” mouse lungs challenged with SARS-CoV-2.
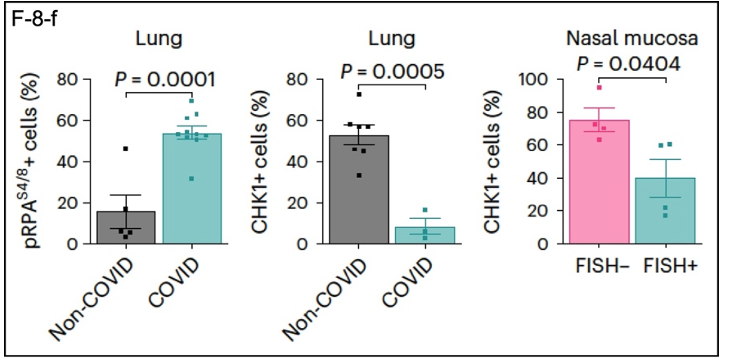
Data from samples of the lungs of COVID-19 patients (F-8-f) are also consistent with the overarching theme of this research. DDR-related proteins show expression patterns that complement those in the other experiments. pRPA is elevated in COVID lung tissue compared to non-COVID tissue, while CHK1 is depleted. A similar result was obtained for CHK1 using nasal mucosal tissue, where SARS-CoV-2 enters the body.
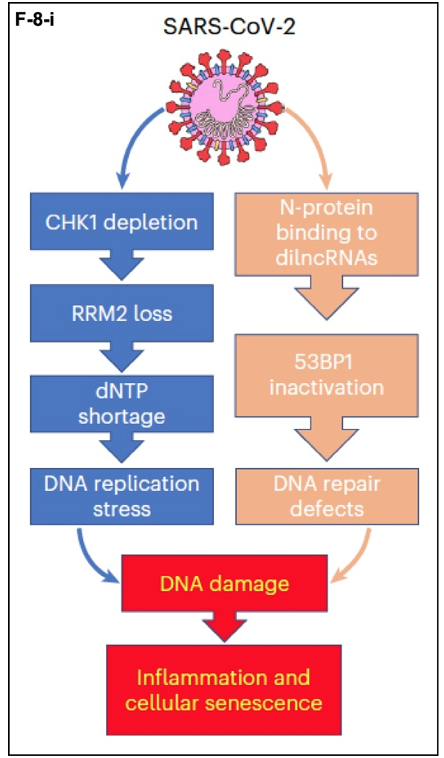
The results of this research can be summarized as follows in (F-8-i). SARS-CoV-2 infects target cells and exerts its pathological effects through two parallel pathways required for maintaining genome integrity. In the mechanism described here, CHK1 depletion leads to depletion of RRM2. The resulting dNTP shortage leads to DNA replication stress and DNA damage. In the parallel pathway, for which the data are also strong, the SARS-CoV-2 N-protein binds to damage-induced, long non-coding RNAs (dilncRNAs). These RNA molecules (7) nucleate DDR complexes at sites where they are required. The viral N-protein disrupts this process and leads to DNA repair defects. The end results are senescence of the infected cells, i.e., they get old and die before their time, and inflammation caused by the death of these cells.
Why in my view is this an exemplary paper? From the ground up the hypothesis is well conceived: SARS-CoV-2 infection dysregulates the DNA Damage Response in individual cells. This is demonstrated in multiple, complementary experiments, only a representative few explained here. The paper then shows that effects on DDR are also present in the animal model represented by the mouse expressing human ACE2. Finally, examination of samples from COVID-19 patients shows that SARS-CoV-2 affects pRPA and CHK1 expression as would be expected based on extensive analyses using cultured cells and the mouse model. From the conclusion to the paper:
Altogether, our results indicate that SARS-CoV-2-induced DNA damage triggers a cell-intrinsic pro-inflammatory programme that, in concert with the immune response, fuels the strong inflammatory response observed in patients with COVID-19. The observed ageing phenotypes recently reported in patients with severe COVID-19 are consistent with our observations. Finally, by proposing a mechanism for the generation of DNA damage and the activation of DDR pathways and of a pro-inflammatory programme, we provide a model to improve our understanding of SARS-CoV-2-induced cellular senescence. In this regard, it will also be interesting to determine if persistent DNA damage and DDR activation, features of cellular senescence, following SARS-CoV-2 infection, contribute to the chronic manifestations of the pathology known as long COVID. (emphasis added)
I see nothing to add to this (8). The results explain much of what is observed in COVID-19 patients, and this research will be useful in understanding long COVID. How this knowledge might lead to effective treatments of COVID-19 is not immediately clear, but a thorough understanding of the molecular and cellular derangements caused by COVID-19 will be essential. As shown in this research, the damage done by SARS-CoV-2 probably resides deep in the cell and can be severe and long lasting for that reason. In the meantime, wear your mask (N95 or better)!
Notes
(1) Within a relatively short time 40 years ago it became clear that HIV causes disease by reducing the population of T-cells of the immune system to levels that prevented the immune system from working properly. This is why AIDS is associated with opportunistic infections (e.g., cytomegalovirus, Pneumocystis jerovicii pneumonia) and other AIDS-associated diseases such as Kaposi sarcoma.
(2) How small is 30,000 base pairs? The coding regions of the two alternative forms of “my favorite protein,” which are on different chromosomes, are ~7,600 base pairs that produce proteins of 2,541 and 2,542 amino acids, respectively. The gene of one version is 27,000 base pairs long when the noncoding introns of the gene are included. The other gene is 300,000 base pairs long, or about 10 times the size of the SARS-CoV-2 genome.
(3) A “cell line” is a term of art denoting a population of immortal cells, i.e., those that do not eventually stop dividing in their “old age” (senescence) in culture; Huh7 cells are immortal cells from a liver tumor. Cell lines are essential for cell biology, but cellular results must be extended to other experimental models for them to be a valid biological result that can be extended to clinical medicine. This has been done in the research described here, which included experiments in mice infected with SARS-CoV-2 and analysis of tissues from human COVID-19 patients. The latter confirmed and extended the results obtained with cultured cells.
(4) pDNA-PK(S2056) = phospho-DNA-PK on the 2056th amino acid in the protein, which is the amino acid serine. Similar designations apply to the other proteins analyzed. These proteins have been separated by size (the 450 kDa marker = a molecular weight of 450,000), transferred to a permanent matrix and identified using antibodies specific for each protein. Immunoblotting is described here if you want do go down that rabbit hole.
(5) RRM2 is a subunit of the essential enzyme ribonucleotide reductase, which converts ribose-based nucleotides (RNA building blocks) to deoxyribose-based nucleotides (dNTPs, DNA building blocks). Without enough dNTPs available DNA synthesis is impaired and affected cells get sick.
(6) ACE2 is usually called the receptor for SARS-CoV-2. “Receptor” has a specific meaning in cell biology. T-cell receptors mediate immune responses. When insulin binds to the insulin receptor, a series of events leads to the clearing of excess glucose from the circulation after a meal. Both of these are normal homeostatic functions in the organism, one necessary for normal immune function and the other necessary for energy metabolism. In my view, ACE2 should more properly be called the binding protein that allows SARS-CoV-2 to enter human cells and cause disease. That the virus binds to ACE2 is an unfortunate evolutionary accident, not unlike CD4, which normally acts as a co-receptor with the T-cell receptor but also binds to HIV and facilitates viral entry into CD4+ cells. AIDS is the result.
(7) Long non-coding RNAs are relatively new to the RNA world of molecular cell biology. Their existence was a surprise, and they are involved in the regulation of gene expression and other processes such as the DNA Damage Response (DDR).
(8) I have at times expressed reservations about a biology paper with 29 authors. This research, however, took that many working scientists to complete in a timely fashion. The project was very large and multifarious. The paper was also under review for 13 months, from December 2021 until January 2023, during which a number of results must have been clarified or confirmed at the behest of the reviewers and editors. Only one author of 29 revealed any possible competing interests, which seem minor in the larger context of the research. The paper is also published in a legacy journal, which might be defined as one that predates the rise, and near dominance, of online “journals.” Nature Cell Biology (1999) qualifies. It has also been very strong in the cell biology community for almost 25 years. But that is not a guarantee. The Lancet (1823) published Andrew Wakefield’s paper on the (nonexistent) relationship between the childhood MMR vaccine and autism. The New England Journal of Medicine (1812) published the paper that confirmed the Pfizer/BioNTech COVID mRNA vaccine is safe and effective.


Absolutely amazing work. Wow! Just curious.
“b. What are the specific dNTP precursors?”
Let’s start with the less-specific ones.
Purines: glycine, glutamine, aspartate
Pyrimidines: glutamine, aspartate
Aspartate for these purposes needs to come from the TCA cycle (a fair reason to enhance TCA and mitochondrial function). Glycine and glutamine can be extrinsic.
More-specific ones come from foods high in nucleic acids (sardines, pinto beans, a variety of other common foods), as discussed extensively by Ben Frank in his books and articles from the 1970s. Quite a bit of literature was published later, in the 1980s and 1990s, on the role of nucleic acid precursors for immunological enhancement. For example:
https://pubmed.ncbi.nlm.nih.gov/12142952/
https://pubmed.ncbi.nlm.nih.gov/8283305/
“c. Are they relatively cheap / available?”
Common foods mentioned are relatively cheap and available, yes.
Glutamine and especially glycine are very cheap. Glycine, glutamine and cysteine are precursors of glutathione, which has general antiviral effect as well as anti-Sars-Cov2 effect. Many other benefits, too numerous to list right now, across multiple pathways and organ systems, including notably (and recently) an apparent anti-aging effect of glycine+cysteine, which is no surprise given the known mechanisms.
Taste is mild. Glycine:glutamine:cysteine in roughly 10:10:1 or 10:5:1 proportion, mixed in water, goes down easy. I speak from extensive experience, e.g. I buy my glycine by the 25-kilo sack. Typical daily intake over many years: 20 grams glycine, 10-15 grams glutamine, 1-2 grams acetylcysteine (common form, probably more effective than native L-cysteine). And oh btw: 3+ years, no vax, no mask (except occasionally when forced), no distancing, and no covid. Though I use other preventives as well (melatonin, etc.).
Great! Thanks. Exactly the info I was looking for.
After reading your post, I decided restock my melatonin supply.
And guess what.
The nanny state, currently determined to drive the UK economy not into a ditch, but into an abyss, as usual, has targeted the most egregious threats to society in its righteous determination to stamp out these frightening perils.
Without warning and nary a word from the public and probably nary a word from scientists with any credibility (we saw during the pandemic seen what kind of scientists the ruling class prefers to consult. An aside, go to Pubmed [not exactly a hotbed of misinformation] and check out the number of positive research papers that have been produced on Ivermectin. I was gobsmacked).
Anyway…
Note::
Melatonin related deaths?
There is however, a problem with children overdosing eating gummy type melatonin, none fatal however..
It is easy, though, to commit suicide with any number of OTC compounds, the solution of which is childproof caps and age restrictions on purchase, not outright bans. And you can still buy all the CBD oil you like in health food stores.
The Latin binomial of soybean is Glycine Max. So there’s that.
Wonder, if this was why C, Rg3, Nicotinomide riboside, quercetin (isoflavones) & NAC looked so promising, at the beginning of the pandemic?
AND: https://www.nature.com/articles/s41598-018-22388-5
I was wondering why I never caught it… Glad they mocked those who suggested these. Really grew my trust in all the narratives out there. The gifts that keep on giving!
You do not know if you have not caught it unless you have had an antibody test. I am in the same boat of thinking I have not gotten it, but I am set up to be able to be pretty strict about avoiding people. Plenty, particularly with Omicron, had asymptomatic cases. But even if you got it, less severe is better!
good point. “I never knowingly caught it.” would be a better claim!
We’d gotten D614.G, alright. BA.1 & a subsequent BA.2xx, since. In March of ’20, we’d had indicative symptoms. But VERY mild, until Hypoxemia ~day 15. So, who knows. I’d used NrPT, zinc, D3. Quercetin, Bromelin & NAC, working 84hr weeks on the road, where we’d no sick-leave, worked in CLOSE proximity to far younger folks (outdoors, in places with infrequently vaccinated kids). We’d read enough Asian epidemiologists’ & NYC HCW’s Tweets to know SARS CoV2’s peculiarities; but PASC & mRNA-1273 side effects were a shock. I’d Mexican & FAR younger coworkers that got 2nd, nastier VOCs before we’d a vaccine a year later. I’ve added Natto/Serra & ALWAYS had a pretty Sirtuin/ phyto-polyphenol rich diet. I still swear by this, after 3yrs of living a veritable 70s disaster movie (the same folks, seem to be proving right, about stuff they’d predict since SARS or HIV?)
Thank you KLG. Indeed inflammatory reactions were known to be associated with COVID pathology in many if not most of its manifestations and this paper shows the reaction has not only to do with impaired immune responses but also the events described here within the cells. This intrinsic pro-inflammatory pathway as they name it. I confess not having read the paper but I have a question for you. It is about this ORF6 of which I don’t know if there if something similar has been described in other CoVs or any other RNA virus or any other gene regardless origin. Could it be hypothesized that if SARS CoV2 losses this ORF6 or is any other way rendered unexpressed the pathology of SARS CoV2 might be much milder?
Please forgive me for my laziness I would like to do it by myself but now I don’t have time to do the effort.
Bravo, KLG! This is an excellent post, not just for explaining the conclusion but for taking lay readers through the evidence.
Given SARS-CoV-2 should be expected to cause premature senescence at the cell level, it will be interesting to see the rates of disease in organs which have “immortal” cell populations rather than the usual more-or-less rapid cell turnover, for example the beta cells of the islets of Langerhans (insulin production / metabolic setpoint regulation in the pancreas), CNS neurones (the brain!), the lens of the eye etc. Will we see increases in metabolic disease and dementias? If we do, it will probably be a long way out in currently young people – perhaps we will see a reduction in the average age of onset of these conditions?
Unsure what you are saying here: ” The Lancet (1823) published Andrew Wakefield’s paper on the (nonexistent) relationship between the childhood MMR vaccine and autism.” In context a double negative? Or are you saying there is no connection between MMR vax and autism
Lancet retracted the Wakefield paper:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2831678/
The paper was based on a study of 12 children, already too small to be valid. And it was cooked to support litigation:
Have we not all learned that so many things in the common narrative may not be as they are presented? I appreciate the research Yves and I share this with all due respect and appreciation for the work you do.
Also not standing up for Wakefield, but isn’t isn’t it somewhat ironic because the justification for removing Dr Andrew Wakefield’s paper was a failure to declare a conflict of interest of £55,000 for a legal consulting fee for a completely different study, when the number of much more conflicted interests are often ignored?
Many studies make contradictory findings and isn’t and wasn’t a valid ground for any retraction, either.
And what about the senior author for the paper, Dr John Walker-Smith, who was exonerated by the British High Court in 2012, which rebuts the Lancet’s claim:
“For the reasons given above, both on general issues and the Lancet paper and in relation to individual children, the panel’s overall conclusion that Professor Walker-Smith was guilty of serious professional misconduct was flawed, in two respects: inadequate and superficial reasoning and, in a number of instances, a wrong conclusion.”
This above criticism was aimed at the General Medical Council, and if they could maliciously use “superficial reasoning” and “a wrong conclusion” to fire one man, then they could have very much done so with Dr Andrew Wakefield. If the study author did not commit “serious professional misconduct” (I.E. his conduct was sound and proper), it means so was his work on the MMR paper.
https://www.independent.co.uk/life-style/health-and-families/health-news/mmr-doctor-john-walkersmith-wins-high-court-appeal-7543114.html
Could surely be wrong, but there is reason to doubt the common narrative.
You are misrepresenting Lancet. The basis for retracting the paper, in layperson terms, was that it was WRONG. Second para from the linked discussion:
The earlier investigation was mainly about the conflict of interest and decided that was not disqualifying.
Great summary of the paper. The only thing I would add is that knocking down these damage-sensing repair pathways, you increase the chances that cancer could develop later on. We will see if Covid ends up increasing the incidence of certain cancers in the future. Let’s hope not.
There is already some evidence that it is, e.g. https://www.sciencedirect.com/science/article/pii/S0165037822002923
I agree that we will have to wait to see how big an effect there is.
I also agree that this is an excellent summary!
Wow even more reason to be taking vitamin D supplementation. It’s know to be important in immunity function support, an inflation moderator AND it is believe to help protect DNA from damage and telomere shortening.
I tried to read this, but I am a dope when it comes to this stuff, but something is bothering me from a common sense perspective.
SARS-CoV-2 only causes disease is some people, many people are asymptomatic. So is this really about the person and not the virus? Are they studying the wrong thing? Like, should they be studying why some people get sick and why some do not?
So it does not make sense to me that we get sick from DNA damage because then everyone who catches the virus would have symptoms.
I don’t know, just thinking out loud.
I believe history has shown that it can take a long time for viral damage to cause symptomatic illness. Just one example
“researchers found that compared with people who had not had a flu infection, those who had the flu had a 70 percent higher risk of Parkinson’s 10 years later, and a 90 percent higher risk 15 years after.”
https://www.nytimes.com/2021/12/20/well/live/flu-parkinsons.html
Assuming the human race has decades left, it may take many of them for people to learn precisely what the SARS Cov-2 virus is doing, and to whom.
So much to learn, so little time!
Not everyone who catches Epstein-Barr virus gets multiple sclerosis.
And what do you mean by asymptomatic? Sometimes the symptoms are so mild, they’re lost in the everyday sniffles, allergies, and tiredness. My own Covid was a mild upper respiratory infection for a few days, mostly post-nasal drip and fatigue. But the effects lasted much longer. I have a sport smart watch, and I could see my cardio fitness drop significantly and stay that way for weeks, if not months.
I feel your reply is a bit antagonistic. I wish we can have an open discussion because maybe I do not know anything. I just feel something is off.
Maybe if I explain it this way; I seem to see that
“Not everyone who catches Epstein-Barr virus gets multiple sclerosis. ”
If we were to make a direct comparison you would see that I agree with that as well. Why though? genetics? Diet? Environment? All three?
“And what do you mean by asymptomatic? ”
Asymptomatic: Neither causing nor exhibiting symptoms of disease. Many people are asymptomatic
So your COVID was not asymptomatic. And most people who catch EBV are asymptomatic.
I think we need to focus on the differences in people and not how SARS2 has some universal way of causing disease. This would be more in line with personalized medicine.
Someone more knowledgable can state this better, but I believe there have been studies that suggest even asymptomatic COVID can cause fairly serious damage. As Carla says, above, it may not be immediately manifest, but could show up in tests. This was posted over a year back, so the data is probably more precise now, but:
I would be curious to hear more on the current data about this, as it seems important to have available for the inevitable run-ins with those who just accept the propaganda.
You are confusing what are called symptoms with actual disease or infection. If you are old enough to remember when Aids / HIV was a major worry/death sentence there were plenty of people who went years being infected with HIV and had no symptoms (AIDS).
It’s like having a couple of drinks and nobody noticing you are actually intoxicated. The lack of noticeable intoxication does not mean your functions aren’t impaired and that you aren’t at risk of causing an accident if you drive or that you could avoid a DWI if pulled over.
Yes! This is exactly my point! And this is how they found treatment for HIV
https://www.youtube.com/watch?v=e6orDqEsfCE
Here is Fauci on it:
https://www.youtube.com/watch?v=uRgtqj3JMKk
But HIV is not COVID, since HIV is a latent virus if latency is achieved.
“Like, should they be studying why some people get sick and why some do not?”
This is a question to which I also wish there were an answer. Analogously, why do only one to three percent of persons infected with polio go on to develop disease? Just a layperson’s guess, but it would seem there are genetic factors at work in such instances.
And thank you, KLG.
I would like to suggest that language about viruses needs to be changed.
A virus is not alive, not a living organism. It does not perform any living functions, left to its own devices, it disintegrates quickly. A virus does NOT do anything, things are done to them.
Consequently, it does not infect anything, it gets ingested, taken in, by a living cell. Inside the cell, cellular processes react to the new protein structure, cleaving it apart. This results in the viral envelope, if present, taken apart first, then when that releases viral payload, the DNA/RNA encoding protein structures are acted upon by the cell DNA/RNA replication processes. In fact, some viral DNA messages are first physically get transported to the cell’s DNA replication complex. Again, no hijacking is forcing the cell to do this, the cell simply does not have the means to differentiate good DNA from bad. RNA processing, such as that of SARS-CoV-2, takes place somewhat similarly.
Questions to note:
1) why do viruses exist with such well defined structure and message format, some like SARS-CoV-2 even have the protective envelope and binding corona protein structure. In fact notice that SARS-CoV-2 can be taken in by a living in two ways: cell’s binding receptor ACE2 wraps around a virus’ binding protein => resulting the cell changing its outer membrane to pull in the object; OR, a virus can simply rub up against the cell’s outer membrane => again the cell pulling virus in as a result {less frequent}.
2) why doesn’t the cell’s DNA/RNA replication + virus assembly from newly made parts ever stop? In fact, cellular death comes from this very fact that after a while too many newly crafted virions inside the cell result in the cell bursting and dying, which we then detect as inflammation. But basically, a cell keeps making new viruses until its resources are exhausted and/or the cell dies.
You said what I felt but could not put to words. It seems like saying “the virus attacks us” was wrong to me. Does a virus have intention?
This is striking to me! Are you saying that our own body creates the disease?
Well, in so much as the disease is the aggregate of symptoms, perhaps.
Inflammation? Broken up cells push and ooze into the surroundings, irritating everyone.
Fever? Elevated tissue temperature breaks down protein structures, yes please!
Headaches? Perhaps vascular restrictions in and around the head indicate blood is needed elsewhere.
Immune response? Send out the hungry dogs to eat the bad wolf, and somebody to remember what they looked like when they come the next time! Done.
One of the bigger mysteries to me is how the immune system knows there’s something to be done about anything. For instance, when cells show signs of an infection event, the immune system takes note. Why? What is the trigger?
I should note I have no degrees or education of any kind, just asking questions. I have not yet figured out the right questions for this last bit.
From what I understand it happens through the neurons.
https://elifesciences.org/articles/66706
What do you think of this link, Emmett? Whether they’re alive or whether they’re machines…no matter how relatively “simple” they are, for nature to have provided such simple phenomena to assist something as complex as new species evolving provides yet more evidence of curiously ideal synergy and
proto-components having been present. To me anyway. https://www.medicalnewstoday.com/articles/host-virus-interactions-have-played-key-role-in-evolution
So, the idea of Horizontal Gene Transfer is indeed a curious one.
Dr. Irwin: “viruses may retain those genes they acquire from their hosts because they are beneficial to the virus. And, for a gene to persist, the organism must survive and propagate, a trait at which viruses are very skilled.”. Now my contention is that viruses do no such thing. They do not survive, nor propagate.
Mutations in the viral genome are introduced by DNA/RNA reproduction facility, which is owned and operated by the living host cell. If such mistakes can result in a whole human gene sequence to be copied into the viral genome, then how does the viral re-assembly work? With this new DNA bit being there? Fair to say I don’t understand how viral re-assembly in the living host cell takes place neither.
DNA repair for living cells happens all day. So, a whole fitting viral genome sequence, or even a partial one, might get copied into a newly built living host DNA by accident(?), if such gene segment happens to be loitering around nearby? Genes may or may code for protein, but if they do and that allows the living host to escape danger or react to other stimuli differently and survive, then yes. An upgraded version of that species is now available to reproduce.
Finally, another bit at the end: “identifying which genes are selected for in viruses can tell you a lot about what process makes the virus more successful, and by extension how it uses its host cell.”
What exactly would make a virus successful? Killing its host fast? Ebola is a winner then, no? Killing the host slowly, but surely? Passing through a host without their immune system taking notice, yet mooching off of most of their resources to make more viruses? Many viruses actually do just that. Again, immune evasion is not something viruses do. If their physical structure has changed, such that the immune system doesn’t recognize them, it’s because some previous host introduced enough changes during their reproduction. Same goes for virulence, if that’s a virus feature at all. My simpleton way of understanding the word virulence, is to make tons of new viruses, fast. These factors are properties of the host cell making the new viruses.
But, interesting ideas. Thanks for the link.
Whoops, out of time to ponder most of what you’ve laid down (thank you; hope sincerely to get back to it).
Only in the case of the female via cord blood, right?
Influenza infection induces host DNA damage and dynamic DNA damage responses
https://europepmc.org/article/PMC/4802977
DNA mismatch repair is required for the host innate response
https://www.nature.com/articles/s41564-019-0509-3
Suppression of RNA recognition
https://www.cell.com/immunity/fulltext/S1074-7613(05)00211-6
So COVID makes your cells get really old really fast. Great news. If they were able to combine this research with the current state of the art of longevity research related to biological aging and tell people that every time you get COVID it is taking something like 6 months to 3 years off your life at a cellular level. I think people would respond differently with their precautions.
Like in the Princess Bride movie where the machine in the dungeon magically removes lifespan by a selected amount. Somebody could make a meme there.
Coronavirus SARS-CoV-2 has a LARGE RNA genome (30,000 RNA nucleotides in one single RNA molecule), small relative to DNA genomes yes, but the LARGEST known RNA genomes.
Relevant beyond parlor knowledge as it was this RNA size and the observation of shorter replication-competent variants (28kb iirc) that allowed for reliably successful packaging of mutated and transgenic viruses for legit research. This, along with differential infectivity and the reconstitution of infectious virus from a small number of cDNA clones excited researchers twenty years ago, when they started mixing matching and passing these 7-8kb clones around like cassettes/doobies at a Dead show, which eventually attracted the attention of all the wrong “private” state actors with a troubling and persistent historical interest in “race”-specific biowarfare agents.
Interesting too that ACE2 polymorphisms across human populations are not publicly curated at this late date.
Minor quibbles Anyhow thanks for the effort. Will read the paper.
Dr. Fauci should win the Nobel Prize for medicine. As a result of his support of enhanced function research he has provided a huge experiment which will certainly advance our scientific knowledge. Sars-Covid-19 has infected perhaps 100s of millions of people and has killed millions. From this massive experiment we are sure to learn many important things about cell biology and its malfunctions. Why work on laboratory animals when we can use massive numbers of humans for this crucial experiment. Dr. Fauci is a brilliant scientist whose support of enhanced function will certainly aid in our further understanding of cell biology and biochemistry. All hail Dr. Fauci for his brilliant contribution to scientific knowledge. He is certainly one of the greatest scientists in the history of humankind.
“It will also be interesting to determine if persistent DNA damage and DDR activation, features of cellular senescence, following SARS-CoV-2 infection, contribute to the chronic manifestations of the pathology known as long COVID.” [from the Post’s source paper]
“As shown in this research, the damage done by SARS-CoV-2 probably resides deep in the cell and can be severe and long lasting for that reason.” [from the conclusion of the Post]
These observations give scary long legs to Lambert’s Corona fears:
“I [Lambert Strether] still think “Something Awful” is coming, however. I mean, besides what we already know about.”
KLM’s post is interesting , complicated and perhaps indicative we should all take an extended course in cellular biochemistry. During the first several months of the pandemic there was limited reporting of detailed pathologic study of victim/ victims of Covid-19. Traditionally the foundation of learning about a new disease, or more about an “old” disease, has been the autopsy. The very early medical and scientific reports on Covid-19 made little sense to me. The article in Lancet 2020; 395: 1417-18 on April 17, 2020 by Vargas et al entitled Endothelial cell infection and endotheliitis in Covid-19 reported the autopsies on three patients. They found viral elements within endothelial cells as well as inflammatory cells with evidence of endothelial and inflammatory cell death. As endothelial cells line all arteries, veins, arterioles, venues, capillaries, the heart and the lymphatic system, the vulnerability of endothelial cells in virtually all organs made sense in the protean organ manifestations of Covid-19 including the brain. Subsequent publications in Lancet and Lancet Haematology in May and July 2020 further suggested an endotheliopathy centered pathophysiology with associated immunologic and inflammatory elements. Another article in the New England Journal of Medicine with similar conclusions was published by Ackerman et al on June 9, 2020.
Elements that still seem missing to me, some of which may not be retrivable would include:
1. Failure to do whole genome analysis on a much larger scale of variants to be able to determine which variants have what clinical features, pathologic, immunologic and intracellular inflammatory changes.
2. Longer followup on those with Covid-19 measuring the above and how long is each variant shed in the stool
3. Population studies to determine characteristics of asymptomatic infected/ carriers
4. Correlate vaccine status with above measurements
5. More intensive monitoring of sewage for Covid-19 and various variants, at least twice monthly by all county health departments
6. Have state DNRs monitor the most common animals with human contact for Covid-19. We already know
Covid-19 has been relatively common in deer in some midwestern states ie IA and MI
7. More basic research in the endothelial cell