By NewdealDemocrat. Originally published at Angry Bear
One of my pet peeves is that economics as a discipline needs to import the entirety of learning theory from psychology, not just parlor tricks like the endowment effect. For example, learning from models.
To wit, once Jack Welch was successful in using a pay scheme at GE that ensured that a given percentage of employees would not get a raise in any given year, it was inevitable that other employers who adopt the idea until it spread throughout corporate America. And it not giving raises to a certain percentage of employees was successful, why not implement it across the board with *all* employees?
Monkey see, monkey do.
As I noted several weeks ago, even though we are at least closing in on full employment, the percentage of employers not raising wages at all has gone up in the last year:
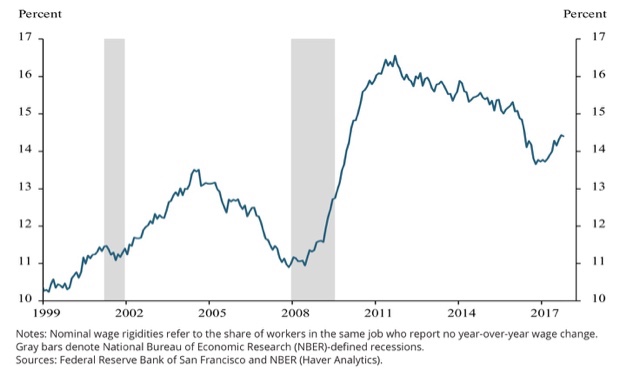
And now, cue Atrios about how big companies, fat with their new tax cut $$$, aren’t planning on raising wages at all:
[E]xecutives of big U.S. companies suggest that the days of most people getting a pay raise are over …. [In] rare, candid and bracing talk from executives atop corporate America, made at a conference Thursday at the Dallas Fed[, t]he message [wa]s that Americans should stop waiting for across-the-board pay hikes coinciding with higher corporate profit …. to cash in, workers will need to shift to higher-skilled jobs that command more income.
….The moderator asked the panel whether there would be broad-based wage gains again. “It’s just not going to happen,” [Troy] Taylor, [CEO of the Coke franchise for Florida,] said. The gains would go mostly to technically-skilled employees, he said. As for a general raise? “Absolutely not in my business,” he said.
This is putting even more deflationary pressure on wages. Since the refinancing spigot has been turned off due to the end of the secular decline in interest rates, if wages don’t increase, exactly where do employers think increased demand is going to come from? Further, if companies freeze wages even during good times, what is going to happen when, inevitably, times turn bad?
Here again are two graphs I have run a number of times already, showing that the YoY% decline in wage growth averages over -2% during recessions:
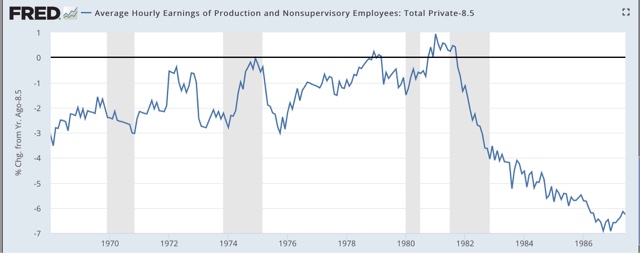
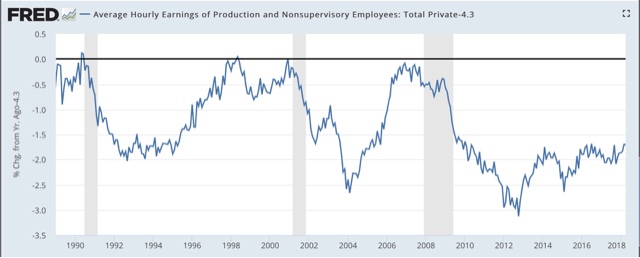
Currently wage growth for nonsupervisory workers is running at 2.6% YoY. Wage freezes are likely to be endemic in the next recession, and even worse, outright wage cuts, potentially leading to a deflationary wage-price spiral, are a significant possibility for the first time in over 80 years.


The rich are waging class warfare on the rest of us. It’s “supply side” economics taken to its logical extreme.
The sad part is that highly paid workers are also very motivated workers who are often likely to more than justify the salary increases. Efficiency wages often are a boon to the employer.
That’s barely the price of the “official” inflation rate. The issue is that there are many people who live in cities where rents go up by 10% on a regular basis. Apart from the highly paid in the upper middle class, like the techies, it is unlikely that their wages can cover that increase in rent alone, unless they got way more than the average. The only others who benefit are those who bought property when it was cheap.
Consider as well that is the average, which means many workers got less. That’s something that is often lost in these statistics – and they get hit even harder by everything getting more expensive.
Often workers have to fight for a measly 2% increase, with many getting no increase, and often seeing job stability go down, which means less income in real terms.
Ultimately the problem is that the economy is oriented to making the rich get richer and screwing the common citizen.
Oops please delete comment. I thought the internet ate my comment so I posted below.
something is up in the skynet, what once was comments obliterating into nowhere has become (on occasion) posted without edit…Check your speling! Oh and good job of rewriting in order to not get labelled spam, you’ve really done two comments on the same topic
Costco just opted to boost their wages:
https://www.seattletimes.com/business/retail/costco-employees-anticipate-benefits-news-with-quarterly-earnings-thursday/
I’m not saying they are perfect, but right now, they seem to be better than most places.
Basically this means that workers are losing ground. Factor in that in many cities are seeing rapidly rising rent and you have a disaster.
http://www.businessinsider.com/rent-cost-growth-in-us-cities-2017-12
Workers have to fight for just a measly 2% raise (2.6% is the average, which means many workers are getting less than that in raises).
Factor in rising rent, food costs, transportation, insurance (For Americans), and other key expenses, then you can see that the real rate of living costs are rising dramatically.
I’m a big believer of efficiency wages – pay workers above market and often the extra motivation, being able to attract top notch candidates, lower turnover, along with other benefits often pays for itself. Sadly, that is not how corporate America thinks.
buy pitchfork futures.The rent keeps going up, the pay keeps going down. I’ll throw my hat in the ring here and say the recessionary forces are working their way down into the general population as the few people who can afford to spend start saving instead, and why wouldn’t they with all the “it was the best of times, and it was the bestest of time…” palaver saturating the airwaves. Just this am I was thinking of adding a fuel surcharge but quickly dismissed that thought as they probably think they pay enough already and they are tightening their belts at the top. Look out below sometime in the next few years at least,I agree with a comment I saw here recently that it feels like 2007 again in a reply to tom stone who had anecdote that prices are falling.. Not gonna be pretty because who will save the rich this time?
Now that the Trump/Russia meme has aged itself to a stinky camambert, I have come to see it as a massive “wag the dog” by the PTB after hillz lost. There was not going to be any discussion about changing the course of the HMS neoliberal thought collective…
Speaking of neoliberals….
Now that I’m living in the country again, I can make zero headway with my neighbors despite agreement on many issues. The problem? An endless supply of legit examples of high profile neolibs insulting the hell out rural folks. And I can’t really argue with them because this is virtually the only ‘liberal’ forum I go to where trash talking poor whites isn’t tolerated. Worse, everytime some conservative bubble yahoo talks civil war, half the lib-o-sphere mocks them and then laughs when you point out that asymmetrical warfare is in fact a thing.
I have gone from only voting part of my ballot to wondering if I should ever vote again. My choices in the MN Democrat primary will be a lifeless corporate stooge appointed to Al Franken’s seat by a neurotic neoliberal governor, or a Bush administration Republican who makes Hillbot hearts throb.
Every part of this country has its problems, but outside our corporate boardrooms I refuse to think any part of the USA is inherently evil or deplorable. But our media keep telling us to hate each other, fight with each other and never, ever unify to throw out the tiny cabal that sucks up all the money and then trash talks the rest of us.
Geographically related to Trump’s electoral ocean of red and the urban isles of blue.
If you only travel by plane, it’s easy to not appreciate that the relatively not-tall Appalachians are a formidable natural barrier/chokepoint for the Northeast. Even today
A relatively modest number of determined people could peacefully halt freight across the mountain passes. A bit l in Brazil last week or France on a semi regular occurrence.
feeding the Northeast via air and sea would be quite interesting to watch. But probably miserable to live thru.
The Sickle of the Hammer and Sickle was the country folk of Russia, a reminder to the urban industrial workers of the Hammer that we are one working class, and not to let the elite divide us.
Neatly, in the post-war West, the empire has done what empires do, and moved populations around their colonies, making the cities into multiple working classes, and the cities and country into really two different-looking populations. I don’t know how to fight that; even a determined effort at solidarity can be broken again by bringing in yet more and different lumpens.
Yes, my experience too. Though I can’t quite figure out why Trump gets a pass on so many things whereas every idiotic thing a D-ish person says or does is proof of nefariousness. I still have to take a deep breath every time I hear someone say Trump “tells it like it is.” Could anything be further from the truth?
I just read a review of a new book about quantum physics
https://bostonreview.net/science-nature-philosophy-religion/tim-maudlin-defeat-reason
that basically argues the controlling narrative in that discipline over the last 90 years has been complete bullsh1t. We know this is so in economics. Look at the narrative that the D’s are trying to sell. Or the R’s. So much BS everywhere you look. The Canadian government just bought a pipeline to ensure tar sands oil gets to market? Ford is going to stop making small cars because they can’t make enough profit on them? What?
We seem to be a civilization in serious decline.
FYI, your link is fubar, the visible text is the URL you want, but the underlying hyperlink is to “https://www.nakedcapitalism.com/2018/06/What%20Is%20Real”, which page does not exist.
Try here.
agreed, tegnost, brilliant diversion. Has bought them 2 years and counting of avoiding talking about the unsustainable, insatiable greed destroying the lives of the bottom 90%.
I believe this is associated with the death of unions. It is easier to get a raise for all employees when the have a union doin their collective bargaining. When you are on your own you become easily replaceable.There are rare exceptions but now companies don’t hesitate to replace people for no reason at all except they can hire someone cheaper. That explains the trend to hiring people and calling them private contractors.
Most unions have been co-opted and are part of the deflationary problem. They set as their initial negotiation targets a 2% wage increase when that doesn’t begin to cover the increase in workers’ costs of living.
Among teachers, the most successful strikes of late have all been “wildcat” strikes. NEA and AFT national and state officials have been doing their damndest to tamp these down, co-opt them, and divert the energy into electing Democrats in the fall. We’ll see how well that works out for them.
Agree. The root of the problem was when general pay raises were replaced with the merit pay system. In my experience merit pay can have too much subjectivity in who got what and occasionally favoritism does occur. Team work in our group over time dissipated and people looked out more for themselves since that was how you got a pay raise. We are not unionized.
What business is that again? Oh yeah, sell diabetes inducing poisonous sugar water, a business that has negative externalities by the truckload. Taylor made it to the top of the Coca Cola slag heap one venal step at a time.
Why anyone would even work for them is beyond my comprehension.
a paycheck
Paying the rent can be a huge incentive in doing crap employment especially if you have dependents.
Lester Thurow wrote years ago about the Japanese system of profit sharing (in a cash bonus–literally–paid in April). Henry Ford and Costco seem to get the need to pay labor for the sake of the proverbial circular flow.
The Fed has screwed up the financial markets so much with its completely skewed access to cheap money that wealth endowments have become largely just a matter of social class. In the next round of pump-and-dump Capital might end up owning everything, with The People owning nothing!
Both R and D parties have been ushering us toward the New Feudalism. Capital Rules! Marx got the diagnosis right, the prescription wrong. Collars on wage contours (as mentioned on ZH this morning) seem reasonable. I like the idea of mandatory profit sharing (using the same metric Management uses to compute their bonuses).
But it will probably take complete collapse (as predicted by Marx) before any reform becomes conceivable, and even then, what it is to change the hard hearts of the elites? Why not just enslave the People in an RFID-chipped social credit prison fed with controlled news feeds and with VR toys that ensures endless compliance?
Neoliberalism has poisoned the minds of the West’s business leaders. Our leaders have no shame as they engorge themselves on fake profits, taking more and more for themselves and leaving less and less for everyone else.
Shouldn’t be surprised that Neutron Jack’s name came up. What I am surprised about is that with hindsight, the massive damage that he did to General Electric is an open book for anybody to read and yet executives are still following his methods and ideas.
In passing – bosses on wages: “No more raises for you!” Maybe the problem is that too many modern executives were brought up on Seinfeld-
“No Soup For You! Come Back 1 Year!”
https://www.youtube.com/watch?v=zOpfsGrNvnk
“t]he message [wa]s that Americans should stop waiting for across-the-board pay hikes coinciding with higher corporate profit ”
In yet another ‘careful what you wish for’ example, If workers expect pay raises to be tied to corporate earnings they should not be too surprised to see their pay oscillate.
it’s not even oscillating, it’s effectively decreasing due to increasing cost of living, as corporate profits rise. oscillation would be a step up.
Yes. Of course oscillation would occur regardless of the larger inflation influenced trend (for which employers are not responsible).
but employers’ profits are increasing, steadily. the wealth keeps getting transferred upwards. empoyers are very much responsible for layoffs and other profit increasing gimmicks, which leave even the workers who survive in increasingly desperate circumstances. this isn’t some inevitable feature of the market. it’s policies designed to have this effect.
Yes, the policies are designed primarily to respond to the requirements of owners and shareholders, not employees. CEOs are pressured above all to keep that first group on board if not happy.
ceo’s run the corporations, not ownerscertainly not small shareholders. the policies are often designed to benefit the managers of the corporation in many cases. the negative effects of our present system are obvious, and not required by anything but the greed of those at the top. as many others have pointed out, in the end it will cost even them.
If that is your belief then your best course of action would be to not participate.
Are you implying that after 2008 people did not take pay cuts? I still haven’t recovered to pre 07 wages, and yes as I mentioned earlier that thought has been on my mind. Corporations share the costs and hoard the profits.
“Corporations share the costs and hoard the profits.”
Very well put.
I spoke with a Walmart employee recently & inquired about wages. (I prefer hearing things first hand, if possible).
He said that having worked there for 20 years (he’s 47), he ‘capped-out’ years ago so would never, ever, get a raise, even if he worked there another 20 years. (No reward for loyalty).
He stated, however, those who were hired in the past couple of years (& were much younger) would make more than him before long. (Another way of pushing out the ‘older’ employees that create higher insurance plans, as those employees must seek other employment because they can no longer live on their same wages of decades ago?)
When I asked if he’d noticed any difference in work ethic among those younger, he was quick to answer that many spent 2 hours or more in their car when going out for a ’15 minute’ break. His disgust was obvious.
I myself have experienced a poor work ethic among many in the younger workforce.
Could it be because they have come to realize that just by showing up they get paid, & will never see rewards for putting forth their best efforts anyway?
(When I experience someone doing their best, I don’t hesitate to tell them how appreciative I am of their efforts, & I’ll even seek out a manager to compliment that fine employee).
The fat cat CEO’s have not only squelched the middle class, but IMNSHO had a direct impact on the work ethics of the generations entering the workforce.
Corporate greed has damaged more than just wages.
Pretty much. Why go the extra mile when you know you’ll never share in any of the gains from that, and the corporation will not hesitate to cut your pay, benefits, and/or hours the very instant it believes it can get away with that, or worse, fire you outright so the CEO can get a higher bonus for this quarter?
I have more or less been running a restaurant for 20 years now. It has gone from a workforce that worked hard but had serious cocaine/alcohol problems, through a wave of xanax users who couldn’t handle any difficulties that came their way, to now barely show up/do anything. I don’t blame these younger folks because the only jobs available are fastfood, dying retail, or uber-eats. Starting your own business is an possibility on par with winning the lottery. They will never own a house, and no one (government/society) cares about them. It seems our society is just running out of energy/momentum.
“Corporations share the costs and hoard the profits”
Corporations share the costs, share the profits, share the risks. There. I fixed it for you.
no, you didn’t.
Something you wrote . . .”if wages don’t increase, exactly where do employers think increased demand is going to come from?” . . . caught my attention.
Why should someone get a raise for just doing their job? If a job is worth X to an employer, why should they pay X-plus? What is the rationale? Has the job being done become worth more? If so, how? And is this linked to that expectation for 2% inflation that has led to a Lincoln that in 1983 cost $20,000 now costing $70,000 35 years later?
My point is, it strikes me like something is wrong to expect across the board pay hikes? After all some folks are diligent about working to better themselves, and climb the ladder of responsibilities to gain a better wage. Others are content to be placeholders. Basically, lacking ambition or drive and posses zero concern for the employer. My question has to do with these folks. Is it your contention they somehow deserve a raise just for breathing, and going to work and doing their job? If so, then it’s small wonder we have inflation that destroys our purchasing power. What is a wonder is that anybody would be motivated to work harder to advance themselves. Are you a closet communist? This last is meant tongue in cheek, but only slightly because your answer has serious implications. I know you support $15/hour, but why not $25 or $50. Think about that carefully.
You need to look at the work of the economist William Baumol, who showed that in a static labor supply situation, it didn’t matter if you personally didn’t produce more year by year; as long as productivity was increasing anywhere else, employers were forced to raise wages to compete against the employers producing more.
For example, you want a violin player? Steel is making more these days, so if you don’t want to lose your violin players to the steel industry in the long term, you need to pay them more to persuade them to keep bowing that fiddle. (obviously in the short term not everyone can immediately retrain)
Of course, immigration reverses this dynamic. You want the same wage as last year, when this new worker says he’ll work for less? Good luck with that!
conversely, why should corporations get increased profits off the increased productivity of their remaining workers, if only due to improvements in technology, while providing increasingly crappy products? why should the ceo’s expect higher salaries for increasing those profits in such circumstances, just for breathing?
Is it your contention they somehow deserve a raise just for breathing, and going to work and doing their job?
Why is this reasoning never applied to CEOs? Why should they reap massive compensation packages while “placeholder” workers deserve nothing?
Why are employers entitled to call the cops if you try to take home the stuff you make at work?
Ok. I’ll pile on. Why should work have any value? Why should a company have to pay for workers at all? What is the value of a given job and who decides that?
I contend that all work has no real monetary value. Just what the market will bear. So it is really just a pricing power relationship, and nothing more.
The Soviet Union adopted this Capitalistic idea of “why pay people anything?” The workers response was “you pretend to pay us? then we pretend to work.” *crumble, crumble, COLLAPSE*
That, and the top down authoritarian centralized control of production and distribution, along with the elites getting benefits like well stocked shops and better quality healthcare and other services. Rather like the United States today.
Regardless of the economic system one chooses, having one that rewards the few while effectively punishing work is a not good for productivity.
One of the original ideas of socialism/communism was to have a bottom up decentralized economic system perhaps using something like a workers’ cooperative in place of a corporation. The government of the Soviet Union started to create smaller decentralized agricultura and industry, but reversed policy which was enforced brutally by Stalin, in part to central power into his hands.
It’s the same reason that authoritarian regimes like the Soviet Union, Nazi Germany, China, and at times the United States Federal, state, and municipal governments do not like independent social, religious, and political organizations. Independent power is threatening. Unions are not being destroyed just because they increase wages, but also create organized individuals into a collective power.
So the supposed system of a government and its economics often aren’t, while the centralization of power and rewards and the problems that that brings is very common.
you do expect yearly increases in the cost of living through the feds purposeful depreciation of the currency in order to keep people from saving too much, right? You do expect your 401k and your corporate stocks to continue increasing in value due to easy money fueling stock buybacks, right? I would suggest you go on amazon order a pitchfork proof vest, cause down in hollywood, you better hope that you don’t run out of gas…
Short answer: because a wage represents a certain purchasing power.
The labor contract is based on an exchange of time for money, yes, but time represents productivity and money represents purchasing power. If you focus only on time and money you miss the more fundamental, and more important relationship between productivity and purchasing power. While the hours and currency exchanged are inherently static, the productivity and purchasing power in the equation are inherently variable. As purchasing power and productivity change, a constant adjustment needs to be made to the static terms of the contract. Since the tendency is for productivity to increase over time (workers become more skilled the longer they stay at a job) and the tendency is for purchasing power to decline over time (inflation is baked into most governments’ monetary policy), there is a rational expectation of continual pay raises.
The other side of “this job is worth X to the employer” is that “this worker produces X for the employer”. The only situations in which “this job is worth X” is meaningful are jobs that any monkey can do; that are so unskilled that no amount of experience ever improves one’s efficiency. Even sweeping the floor can be done poorly at first and better and more efficiently as the person gains experience doing it! I think the meme “this job is worth X” only has currency because of robotics. A robot, typically, does not increase efficiency over time. Therefore robots should not expect wage increases. But everybody else…
I’ve never seen an automation effort – robotics, phone systems, point of sale – that didn’t involve ongoing and increasing costs over time. Maybe the next big cost cutting measure will be forgoing maintenance contract renewals and eliminating security patches on technology. Fingers crossed all around
Next?
Your comments are typical Conservative elitist thinking. Conservatives view all others as “immoral, lazy, unworthy sub-humans to exploit and oppress.” In truth, the laziest, immoral people are all at the top. Owners/Executives sit around smugly with their feet up on the desk, hitting keystrokes for data (worked on by someone else), signing legalistic BS documents (prepared by someone else) getting 6,7,8 figure salaries they don’t deserve for products done by other people.
Why should the worker be subservient to the employer? Citizens owe NO LOYALTY, moral or legal, to a someone else’s money making enterprise. And that enterprise is strictly a product of signed commercial legal documents. Commercial enterprise has no natural existence. It is a man-made creation, and is a “privilege”, not a “right”; just as a drivers license is a privilege and not an absolute right.
In truth the employer/employee relationship is one of mutual benefit derived from different skills. Equality and reciprocity is the heart of every relationship. Conservatives of all stripes (capitalist, communist, fascist, libertarian or whatever else they call themselves) reject equality and reciprocity.
They believe totalitarian ideas of order, submission, divide-and rule, and power for social elites at all costs. They cannot tolerate anyone else deriving equal benefit: to them it is a moral outrage against the (undeserved) social status of Conservatives/Capitalists/whatever else they call themselves.
Capitalism is the religion of avarice, the gospel of arrogance and the moral force of tyranny. It’s purpose is concentration of power and the spreading of misery.
Well Said Buckeye.
In aggregate, wages = demand in a consumer economy: the money company A pays its workers is how workers buy goods/services from companies B through Z. Sure, company A alone can freeze to improve profits, but only at the cost of (slightly) decreasing demand for goods/services from the other companies. If all companies freeze wages, then demand goes down for all companies b/c workers don’t have $$ to buy good/services, and likewise profit.
As for whether workers “deserve” an increase: By performing their jobs, workers produce value for the company. If a company is profitable, workers should get a cut, and if profits go up, so should their cut. If a particular individual isn’t performing well then in an efficient/well-managed company they’ll be replaced; if the job itself is not structured to produce well then in an efficient/well-managed company the job will change; and if the company fails to do either well then in an efficient/well-managed economy the company will change/be replaced.
A $15 minimum wage — and subsequent increases that are ideally tied to some measure of inflation — protects workers and employers from the depredations of companies that would seek to profit (at the expense of reducing demand in the overall economy) by cutting wages. And in that way, it protects all of us from a deflationary spiral.
As for the slippery slope (why not $25 or $50/hour), that’s a great reason why CEO pay should be slashed.
Most commenters here have addressed the wage issue in the context of adding to a company’s productivity and whether they are worthy of a wage increase compared to the CEO’s compensation. What is not being addressed is the cost of real estate and rents taking ever larger percentage of take-home pay and rent increases being a large driver of inflation.
Having gone through a period of unemployment recently it was demoralizing to see that employment opportunities for science graduates starting at $14 and $15 per hour in cities where rents are between $1800 and 2400/month. Many of the younger grads have student debt to contend with as well. How many of these underpaid and rent-gouged new hires who have corporate 401K benefits realize that they are investing in multinational real estate development companies that are promising 4%, 5%, 6% ROI, while their wages remain flat. They may inhabit apartments owned by the same corporation and pay 60-75% of their wage in rent.
Natural selection has generally worked to eliminate parasites that kill their hosts too fast – at least before they have the opportunity to reproduce. I’m not sure the corporate real estate rentiers are following this rule.
John Beech, all wrong! You suffer from thinking that you are at all different from, not only other humans, but just plain dirt. Work harder!! And, I don’t mean at your job, I mean getting in touch with “yourself.”
The rising tide of productivity has raised a lot of yachts. The rowboats the rest are in are weighed down by a heavy anchor, and all we want is a few more links in the rode so we don’t drown.
Anyhow, heard the latest about the greatest? Jeff Immelt, the CEO that took GE over after the plastic pellet salesman CEO mentioned above, earned millions and millions and millions, yet his executive decisions destroyed billions and billions and billions of dollars. Was he paid what he was worth?
tl;dr the propaganda has worked on Jon Beech
It’s weird so many people are against unions and worker organization yet do not see the contradiction of every other aspect of economic life being organized in industry bodies, trade associations, commerce chambers, lobbying groups, special interest groups, think tanks.
It seems businesses understand perfectly well the logic of association to protect and further their interests but lose this clarity when it comes to workers.
Even if you consider most people do not have time for history most of the anti-union sentiment dwells on extremely simplistic narratives of corruption as if having one bad meal or one bad bank discredits the whole idea of restaurants or banks. It’s simply not a logical reasoned position.
It’s interesting to see how pervasive and systemic propaganda in the media and special interest groups can turn people against their own self interest.
Ultimately the neoliberal ideology of ‘wealth creators’ and ‘takers’ must be thoroughly engaged and debunked. Often toxic ideologies like eugenics, racism and this kind of brazenly self serving neo-feudalism are not properly engaged and are allowed to fester on. They thrive behind the scenes and the result is our modern system that has no place for basic humanity or even dreams of a better society and celebrates and rewards unethical behavior and naked greed.
Agree absolutely about vehemently and effectively deligetimizing toxic ideologies. At the top of the list should be the Protestant Work Ethic. All it has done is tie a person’s worth to their wealth, turned life into an endless treadmill of labor, and justified exploiting anyone considered “lesser”. The Reformation/Early Modern period replaced “Divine Right of Kings” with “Divine Right of Wealth”. Where once a very small group of nobles ran everything but were dependent upon the serfs for their own survival, now we have a large group of wealthy despots who can shift their power around and aren’t tied to one local area.
As Greenspan said in 1997 in typical Alan-speak:
Chomsky translation:
and the only change since is that insecurity has increased
What bullshit. The people who get raises are those with the most political capital, not the most skilled. These people are on another planet.
More like comical bullshit.
Boycott Coca Cola, pinch a penny out of Warren Buffet’s pocket.
People are not paid what they’re worth, they’re paid what they manage to bargain for. Some (most) economists believe that whatever a person is able to bargain for that is what the person is worth….. As approximations done by economists goes it is not the worst approximation made but that doesn’t say much :-p
Remove the bargaining power and see what happens to wages…. Or don’t, the decline in unions has already been accomplished so we can easily see what happens when bargaining power disappears.
There was a claim that increased wages would lead to increased inflation.Yep, it is true but for the majority of workers the difference would be so small that it most would almost impossible to measure. How much more would a cup of coffee cost if wages of the person making the coffee increased by 50%? How much of the sales price of a car made by GM goes to pay for the CEO of GM? Does he get one dollar per car? More? Less? If adjuncts at universities got a wage increase of 5% how much would the cost of tuition need to increase to cover that?
Just a small reminder: the CEO of GM is “she.”
Re: “closing in on full employment,” there’s always this:
https://www.zerohedge.com/news/2018-06-01/record-959-million-americans-are-no-longer-labor-force
Of course, the link is missing a decimal point after “95”
When the monthly jobs report comes out, and the media crows about how wonderful the economy is, they never tell us if the jobs are full-time or part-time, if the jobs are low-paying, and what industries the jobs are in. They never mention it takes a minimum of 150,000 jobs created just to keep pace with population growth. And they never mention that one-third of the workforce is still out of work. But anybody still breathing just has to look around to know better.
A piece on medium.com recently concluded that America is innovating a new kind of poverty. One where workers appear statistically to be making an average living but are really only scraping by. The preferred focus is on the statistics of “jobs” and “earnings” — all defined to reflect policy success — as proof Americans are doing ok, without any acknowledgement of the context and conditions workers’ well-being and income are being subjected to in the extractive,predatory economy.
So appropriately in context, even on an average income it’s possible to not be able to afford some of these: healthcare, housing, transportation and food — not to mention education/university for the kids. Especially in expensive cities.
This ‘innovation’ sure does enable a sidestep of reality by the neoliberals. And the neoliberals do not care about people in this position, as long as they can convince those who they deem to matter that everything is great in America (that’s you Trump, Hillary and Obama) with the “lies, damn lies, and ..statistics.”
If the American system of capitalism is so great, why are so many people who work and support it so desperate?
The FDR America is what people want. Thing about FDR was he knew how to save the system. Since 2008 the cost of shelter where there are jobs did not go down but went up.
The old landlords raise rents and the new landlords raise rents. Wages don’t go up so more of what the worker has to work with goes towards a place to live.
The crime of living someplace too long goes up.
How much longer before there is revolt? What good is that great job in San Francisco or DC if rent takes all your money?
P.S. If you know where the rent is going down and the wages are going up, we need to know about it.
Thanks
Here in the Bay Area both are, but with the rent rising faster, of course.
Maximising profit requires minimising wages, it is the inherent class struggle within capitalism.
Neoclassical economics focuses on capital accumulation and maximising profit.
What happens?
Mariner Eccles, FED chair 1934 – 48, passes comment the last time they used neoclassical economics in the US in the 1920s.
“a giant suction pump had by 1929 to 1930 drawn into a few hands an increasing proportion of currently produced wealth. This served then as capital accumulations. But by taking purchasing power out of the hands of mass consumers, the savers denied themselves the kind of effective demand for their products which would justify reinvestment of the capital accumulation in new plants. In consequence as in a poker game where the chips were concentrated in fewer and fewer hands, the other fellows could stay in the game only by borrowing. When the credit ran out, the game stopped”
This time it’s global.
2014 – “85 richest people as wealthy as poorest half of the world”
2016 – “Richest 62 people as wealthy as half of world’s population”
2017 – World’s eight richest people have same wealth as poorest 50%
Look at the returns on capital; they are low/negative because there is no demand for it.
Look at inflation; it is so low because of the subdued demand.
The system falls over when the wealthy take too big a slice of the pie.
In the present day of globalization, American labor must be crushed to a world level playing field in order for those businesses to compete on the same world level playing field. And, we all know how that part of globalization is working. By not increasing labor’s purchasing power in a timely manner labor is pushed to borrow more deeply for the necessities…food and shelter; transportation; higher education; medical care and so much more. Only when the masses of crushed labor become more socially conscious and recognize the role labor has historically played in helping make this country wealthy will the battle again be taken up to re-organize that labor and claim labor’s share of that wealth. Until that time comes we will spiral downhill.
The working class’s greatest weapon: Malicious Obedience.
That is: doing what the Boss tells you to do, even when it is the worst plan possible.
You only get pay raises by jumping ship and by a lot. The tech market is so hot, companies are offering younger employees my salary (I have 15 years exp) plus stock options that can be worth a lot in the future.
I know one of these guys and his story is a riot. He’s currently working at a big company (not FAANG) and he’s struggling there. However he goes to these meetups where people would practice and discuss programming interview questions. After 4 months, he’s good enough to pass all sorts of interviews and that’s how he scored his new well paying job.
There’s definitely an inflationary wage pressure but only if you are young and in tech. Otherwise, it’s a race to the bottom.
Our brightest minds are focussed on making money.
What is money?
It has little intrinsic value in itself; it is the things money can buy that give it value.
Where is the real wealth of nations?
The 1600s showed the Dutch tulip bulb market wasn’t a store of real wealth.
The 1920s showed the US stock market wasn’t a store of real wealth.
The 1980s showed the Japanese real estate market wasn’t a store of real wealth.
The 1990s showed the US stock market wasn’t a store of real wealth.
The 2000s showed the US real estate market wasn’t a store of real wealth.
GDP is real wealth; the wealth in the markets has a habit of evaporating almost over-night.
Why did they invent the GDP measure?
Markets were all the rage in the 1920s and they thought the US stock market represented real economic activity, but in 1929 they found it didn’t.
“Stocks have reached what looks like a permanently high plateau.” Irving Fisher 1929.
Neoclassical economist, who believed in the markets, stable equilibriums, rational market participants and all the rest of that nonsense.
The holy trinity of economics are supply, demand and the money supply, which should all rise together in a perfectly growing economy. This is what GDP effectively measures and any one of the three can limit growth. Money is just the thing that allows the goods and services in the economy to be purchased. Too much and you get inflation and too little and you limit demand in the economy.
Let’s concentrate on making money, profit and capital accumulation with neoclassical economics.
What happens?
Mariner Eccles, FED chair 1934 – 48, passes comment the last time they used neoclassical economics in the US in the 1920s.
“a giant suction pump had by 1929 to 1930 drawn into a few hands an increasing proportion of currently produced wealth. This served then as capital accumulations. But by taking purchasing power out of the hands of mass consumers, the savers denied themselves the kind of effective demand for their products which would justify reinvestment of the capital accumulation in new plants. In consequence as in a poker game where the chips were concentrated in fewer and fewer hands, the other fellows could stay in the game only by borrowing. When the credit ran out, the game stopped”
This time it’s global.
2014 – “85 richest people as wealthy as poorest half of the world”
2016 – “Richest 62 people as wealthy as half of world’s population”
2017 – World’s eight richest people have same wealth as poorest 50%
Look at the returns on capital; they are low/negative because there is no demand for it.
Look at inflation; it is so low because of the subdued demand.
The system falls over when the wealthy take too large a slice of the pie.
We have forgotten all the lessons they learnt the last time they used neoclassical economics.
The greatest disease ever afflicting the human race is a global pandemic with is deepest roots in London. New York, and Washington DC.
It’s called Greed.
Where the article quotes “Atrios,” it looks like it should be “Axios.” :)